
In der Regel beginnt die Prozessvalidierung damit, dass der Chef möchte, dass etwas validiert wird. Doch wie geht man nun am besten vor?

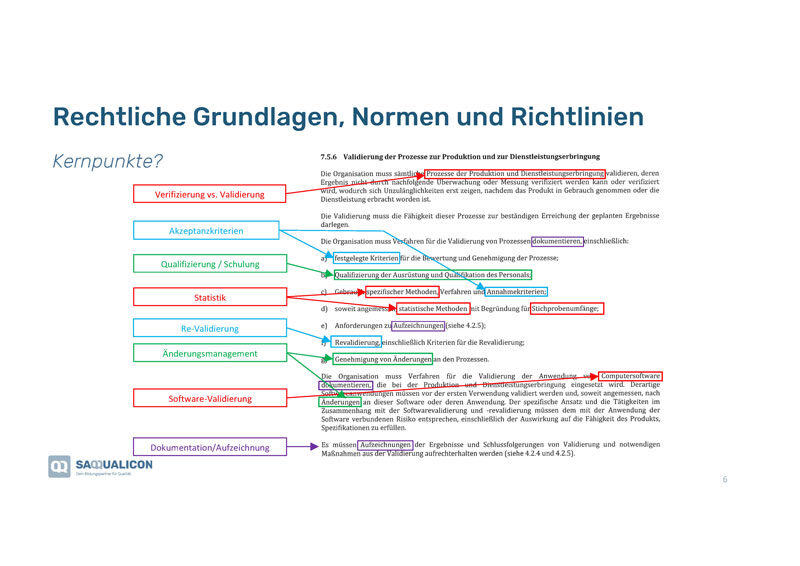
Boris zeigte uns, was relevant ist. Hierfür schauten wir uns zuerst die europäischen regulatorischen Vorgaben der EN ISO 13485:2016, Abs 7.5.6, «Validierung der Prozesse zur Produktion und Dienstleistungserbringung an»:
- Zuerst ist abzuklären, was genau zu verifizieren bzw. validieren ist.
- Akzeptanzkriterien eruieren und festlegen.
- Alle in der Prozessvalidierung involvierten Personen sollten geschult sein.
- Statistisches Know-how muss vorhanden sein.
- Es sollten Regeln für die Re-Validierung erstellt werden.
- Für Änderungen müssen Regeln definiert werden, wer wann was wie ändern darf und wie das dokumentiert werden soll.
- Auch die Software, die für den Prozess im Einsatz ist, muss validiert werden.
- Alle Ergebnisse und Schlussfolgerungen sind aufzuzeichnen und die notwendigen Massnahmen müssen dokumentiert werden.
Wie schaut das in den Vereinigten Staaten von Amerika aus?

Bei den FDA-Forderungen 21 CFR Part. 820, Sec. 820.75 «Process validation», werden praktisch die gleichen Punkte verlangt:
- Es ist festzulegen, was genau zu verifizieren bzw. validieren ist.
- Es werden statistische Verfahren verlangt.
- Die eingesetzte Ausrüstung muss fachmännisch sein.
- Sie verlangen eine Überwachung des Prozesses.
- Die Akzeptanzkriterien sind zu definieren.
- Neben der qualifizierten Ausrüstung wird ausserdem qualifiziertes Personal verlangt.
- Regeln für Änderungen bzw. ein Änderungsmanagement werden auch hier gefordert.
- Die Re-Validierung muss festgelegt werden.
Fazit: Die ISO- und FDA-Forderungen sind praktisch gleich.
Begrifflichkeiten
Bevor wir nun mit der Prozessvalidierung loslegen können, müssen wir zuerst die Begrifflichkeiten klären:
Bei «Verifizieren» ist «(über)prüfen» gemeint, beim «Validieren» hingegen wird sichergestellt, dass ein geeigneter Prozess (Prozessparameter) innerhalb definierter Grenzen (Akzeptanzkriterien) zu einem vorab definierten Ergebnis führt. Hierzu zwei Beispiele aus dem Alltag:
Verifizierung: Ich verkoste die fertige Suppe, ob diese schmackhaft ist.
(Prozess-)Validierung: Ich beurteile (validiere) den Prozess des Suppenkochens anhand der Zutatenliste und dem Rezept, ob diese stimmig sind und ich dadurch als Resultat eine schmackhafte Suppe erhalten würde. Nicht valide könne zum Beispiel sein, wenn ich für ein Liter Wasser ein Kilogramm Salz verwenden würde.
Nutzen
Der Nutzen der Prozessvalidierung ist vielfältig, auch wenn man keine rechtlichen Normen einhalten muss:
- Erfüllung der normativen und gesetzlichen Vorgaben
• Öffnet neue Märkte
- Hohes Prozess-Know-how
• Optimierungsmöglichkeiten
- Erhöhte Prozessstabilität / Verringerte Prozessvariabilität
• Möglichkeiten zur Automatisierung
• Weniger Ausschuss
• Weniger Verschleiss, längere Wartungsintervalle
- Tool oder Ergebnis für die Entscheidungsfindung zur Produktionsfreigabe
- Weniger Abhängigkeit von In-Prozess- und End-Prüfungen
• Prüfumfang kann reduziert werden
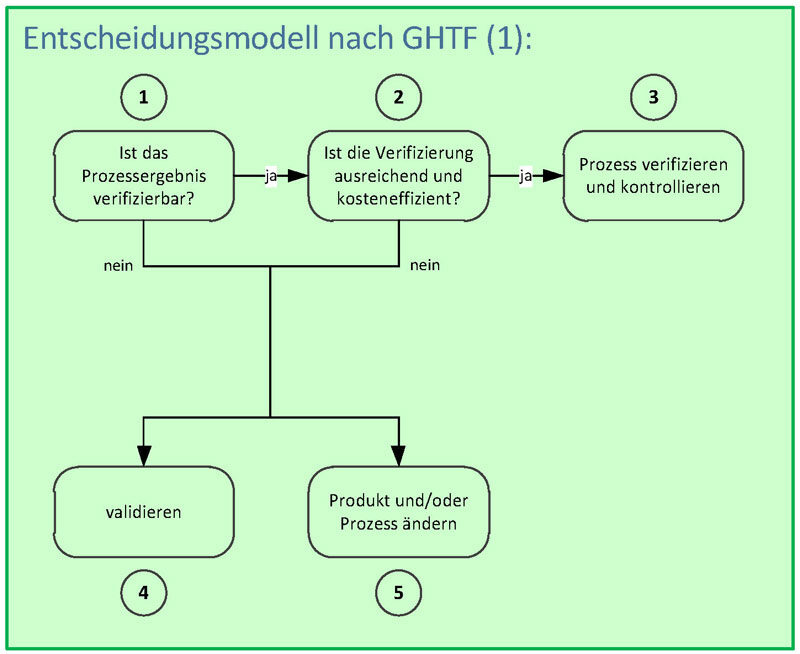
Prozessvalidierung – validierungspflichtige Prozesse
Wie findest Du nun heraus, ob Du einen Prozess überhaupt validieren kannst und solltest? Hierzu gibt es ein hilfreiches Entscheidungsmodell der Globalisation Harmonisation Task Force (GHTF), das Du in der Grafik rechts siehst:
Schritt 2: Bewertung, ob eine Verifizierung ausreichend ist bzw. inakzeptable Risiken bleiben und der Aufwand dafür kosteneffizient ist.
Schritt 3: Prozess wird verifiziert und kontrolliert, sofern Schritt 1 und 2 mit «ja» beantwortet werden kann.
Schritt 4: Wird Schritt 1 oder 2 mit «nein» beantwortet, muss zwingend validiert werden.
Schritt 5: Eine Produkt- oder Prozessänderung kann eine Möglichkeit zur Prozessstabilisierung oder -verifizierung sein.
Validierungsteam
Validierungen können nicht seriös als Einzelpersonen durchgeführt werden. Hier für sind Teams nötig, die die folgenden Kriterien erfüllen:
- Funktionsübergreifend tätig
- Mit Erfahrung und Know-how aus allen Bereichen, die den Prozess betreffen, wie QS, QM, Ingenieurwesen, Produktion und evtl. Forschung, Entwicklung, Beschaffung, Planung
- Festgelegte Verantwortungen und Befugnisse

Neben dem Team benötigt es auch einen Validierungsmasterplan (VMP). Der VMP ist das übergeordnete Qualifizierungsdokument, in dem alle Qualifizierungs- und Validierungsaktivitäten beschrieben werden. Kurz: Dort ist alles drin, was in der Norm steht.
Die Bedeutung der Prozessentwicklung in der Prozessvalidierung
Gemäss FDA hängt der Erfolg der Prozessvalidierung von den erarbeitenden Informationen aus der Produkt- und Prozessentwicklung ab. Erkenntnisse daraus sind:
- Einflussfaktoren bzw. Ursachen für Schwankungen
- Möglichkeiten des Erkennens von Schwankungen und deren Ausmass
- Verständnis der Auswirkung der Schwankungen auf den Prozess und die Produkteigenschaften
- Möglichkeiten für Kontrollmassnahmen
Wer einen Prozess nicht kennt, kann diesen dementsprechend nicht validieren. Das heisst, man muss sich das Wissen selbst aneignen oder die Personen mit dem entsprechenden Know-how ins Validierungsteam holen.
Eine Möglichkeit der Prozessentwicklung ist die «Design of Experiments»-Methode (DoE). DoE ist eine Methode des Qualitätsmanagement-Systems Six Sigma mit dem Ziel, den Aufwand für Versuche zu reduzieren, mit denen die Wechselwirkungen untersuchter Parameter bestimmt werden.
DoE wurde explizit dafür entwickelt, den Einfluss von vielen Faktoren in einem System möglichst schnell und strukturiert zu analysieren. Mit DoE ist es möglich, eine nahezu beliebige Anzahl von Faktoren auf ihre Wirkung für mehrere Zielgrössen zu untersuchen und eignet sich daher ideal für die Prozessentwicklung.
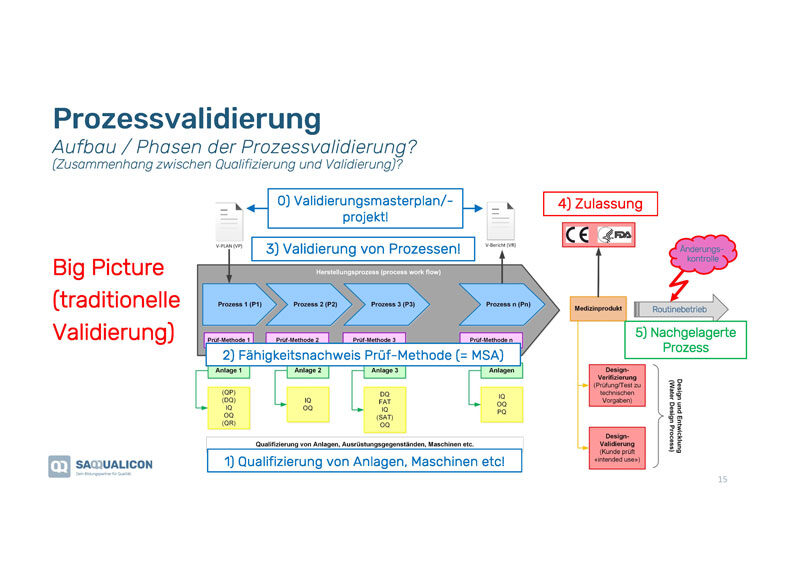
Das «Big Picture» der Prozessvalidierung
Bei der Prozessvalidierung handelt es sich immer um Prozessreihen wie Du in der Grafik nebenan siehst. Bei den Phasen der Prozessvalidierung ist der Zusammenhang zwischen Qualifizierung und Validierung wichtig:
1. Qualifizierung von Anlagen, Maschinen etc., die in dem Validierungsprozess überprüft werden.
2. Fähigkeitsnachweis der Prüfmethode (=MSA), damit diese valide ist bzw. funktioniert.
3. Validierung der einzelnen Prozesse in der Prozessreihe.
4. Validierung der Zulassung (normabhängig).
5. Prüfung des nachgelagerten Prozesses, damit dieser mit dem Output der vorgelagerten Prozessreihe ebenfalls funktioniert.
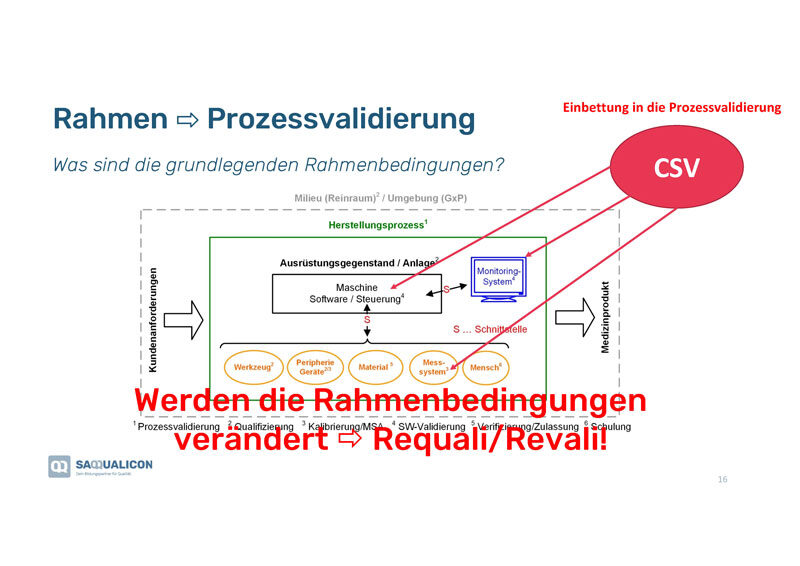
Rahmenbedingungen bei der Prozessvalidierung
Es ist wichtig, immer die grundlegenden Rahmenbedingungen in Bezug auf die verschiedenen Komponenten, wie z.B. Werkzeuge, Materialien, Messsysteme usw., und deren Schnittstellen (in der Grafik mit dem «s» gekennzeichnet) festzulegen und entsprechend zu dokumentieren. Wenn diese verändert werden, kommt es in der Regel zu einer Re-Validierung, da veränderte Rahmenbedingungen das Validierungsresultat beeinflussen. Die Veränderungen sollten unbedingt dokumentiert werden.
Standard Operating Procedure (SOP) bei der Prozessvalidierung
Zu den Rahmenbedingungen gehört auch die Prozessbeschreibung. Sogenannte SOPs sind die Beschreibung der Standardvorgehensweise für die einzelnen Prozesse. Dabei werden die folgenden Fragen beantwortet:
- Warum mache ich das?
- Was ist das Ziel / gewünschte Prozessergebnis?
- Welche Maschinen und Ausrüstungsgegenstände sind involviert?
- Was sind die Prozessmerkmale?
- Wie läuft der Prozess ab?
- Was sind die Rahmenbedingungen?
Eine detaillierte Prozessbeschreibung ist wichtig: Alle müssen wissen, um was es geht und die Rahmenbedingungen (bspw. In- und Output) kennen. Und das von Anfang an – bereits bei der Planung.
Provokatives Fazit von Boris: Was ist nicht beschreiben kann, kann ich auch nicht validieren!
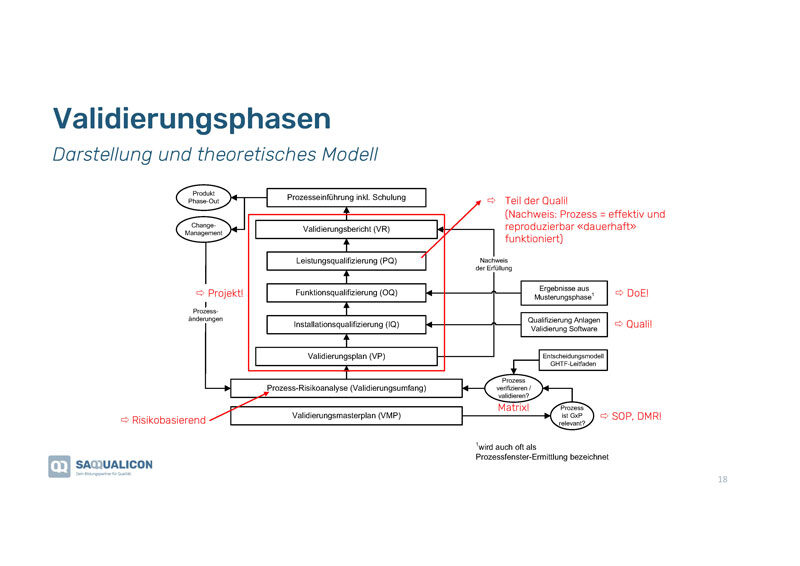
Validierungsphasen – Darstellung eines theoretischen Modells
Zuerst muss man sich fragen, ob die Validierung einen Einfluss auf das Produkt hat. Falls nicht, muss man erst gar nicht damit beginnen.
In der Grafik siehst Du schematisch dargestellt die Validierungsphasen, die zu durchlaufen sind.
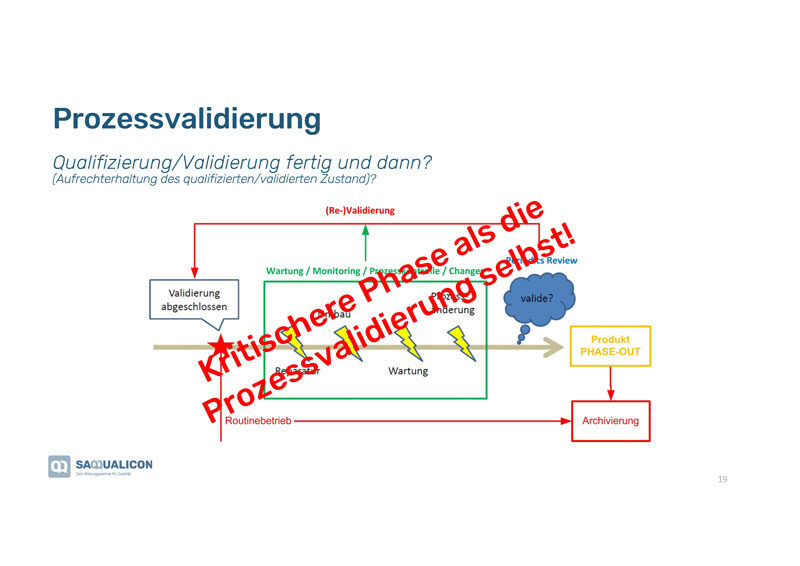
Der Validierungsprozess konnte erfolgreich abgeschlossen werden, alles ist dokumentiert und nun lehnt man sich zurück – die Routine kommt. Doch jetzt lauern die grössten Gefahren:
- Reparaturen
- Umbauten
- Wartungen
- Prozessanpassungen
- etc.
Die häufigsten Fehler
- Fehlende Strategie
- Keine oder unzureichende Bildung von Produktfamilien
- Einzelkämpfer (kein Team)
- Unzureichende oder unvollständige QM-Vorgaben
- Unzureichende oder keine Berücksichtigung der normativen Vorgaben
- Unzureichende oder fehlende Spezifikation / kritische Merkmale
- Keine brauchbaren kritischen Maschinen Parameter
- Unzureichende Berücksichtigung von Erfahrungen
- Falsche Priorisierung / Fokussierung der Risikoanalysen
- Unzureichendes oder fehlendes Prozessmonitoring
- Ohne Plan voran
- etc.
Fazit von Boris: Eine schlecht geplante und schlecht konstruierte Anlage oder ein untauglicher Prozess wird durch eine Qualifizierung / Validierung nicht besser… Hier hilft auch keine Prozessvalidierung!
Nun folgte der zweite Teil mit Camilla Streuli, Stv. Leiterin Lebensmittelsicherheit bei der Delica AG mit einem Beispiel aus dem Lebensmittelbereich:
In der Lebensmittelindustrie ist man verpflichtet, ein Lebensmittelsicherheitskonzept zu integrieren. Delica AG ist deshalb IFS 8.0 zertifiziert.
Sie haben ein Qualitätsmanagementsystem, das alle Prozesse abbildet. Teil davon ist ein Hygienekonzept, die Gefahrenanalyse wie auch das Rückverfolgbarkeits- und Rückrufkonzept. Alles zusammen bildet das Lebensmittelsicherheitskonzept der Delica AG, das die Basis für die IFS 8.0 Validierung des HACCP-Plans und die Festlegung von Verifizierungsverfahren ist.
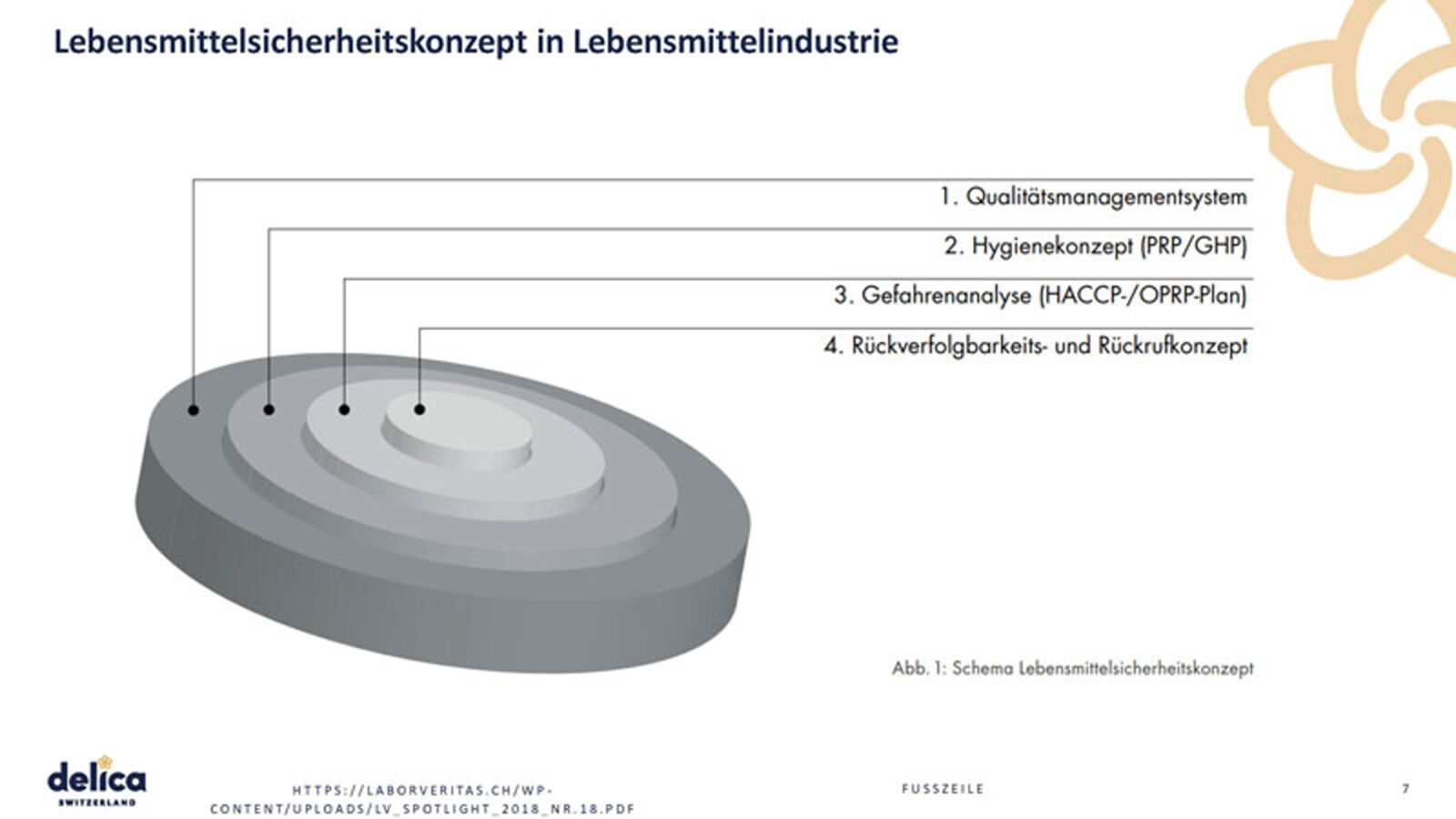



Präventiv-Programme – Konzept PRP
In der Delica AG bestehen sehr viele unterschiedliche Konzepte, die präventiv greifen. Vom Food Fraud (Inverkehrbringen von Lebensmitteln mit dem Ziel, durch vorsätzliche Täuschung einen finanziellen oder wirtschaftlichen Vorteil zu erlangen) über Food Defense (Produktschutz von Lebensmitteln vor geplanter, vorsätzlicher Kontamination oder Verfälschung durch biologische, chemische, physikalische oder radioaktive Substanzen im Rahmen eines Sabotageakts oder eines terroristischen Angriffs) bis hin zur Lagerung/Transport der Lebensmittel.

HACCP Konzept
Unter der Abkürzung HACCP verbirgt sich die englischsprachige Bezeichnung «Hazard Analysis Critical Control Points». Die deutsche Definition von HACCP lautet «Risiko-Analyse Kritischer Kontroll-Punkte». Das Einhalten des HACCP-Konzepts stellt sicher, dass Konsumenten und Mitarbeitende vor Keimen und Krankheitserregern, die beim Arbeiten mit Lebensmitteln entstehen können, geschützt werden – vom Einkauf bis und mit Auslieferung des fertigen Produkts.
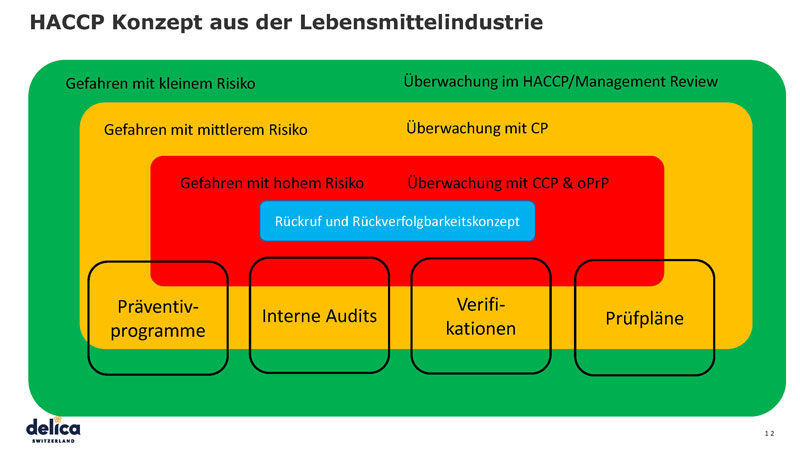
Das HACCP-Konzept ist die Basis für die Klassifizierung der weiteren Prozesse in die nachfolgenden Kategorien:
- Gefahren mit kleinem Risiko
- Gefahren mit mittlerem Risiko
- Gefahren mit hohem Risiko
Überwacht wird alles mit dem HACCP/Management Review. Bei mittleren Risiken wird es mit Kontrollpunkten (CP) und bei hohen Risiken mit kritischen Kontrollpunkten (CCP) oder operativen präventiv Programmen (oPrP) überwacht.
Überprüft wird alles mit Präventivprogrammen, internen Audits, Verifikationen und Prüfplänen. Die Validierung wird über alle Prozesse hinweg gemacht.
Das Rückruf- und Rückverfolgbarkeitskonzept ist quasi die «Lebensversicherung», falls doch einmal etwas schief gehen sollte.
Begrifflichkeiten bei Delica AG
Gemäss Camilla hat Delica die Validierung und Verifizierung folgendermassen definiert:
- Validierung des Lebensmittelsicherheitssystems: Are we doing the right things?
- Verifikation des Lebensmittelsicherheitssystems: Are we doing the things right?

Gefahren von Fremdkörper in Lebensmittel
Wenn man die Bilder auf der Folie anschaut: Das möchte niemand in Lebensmittel haben! Dennoch sind das alles reale Gefahren bei der Lebensmittelproduktion:
- Schrauben von Produktionsmaschinen
- Schalenteile von Nüssen (Zahnschäden)
- Metallspäne von den Produktionsmaschinen
- Büroklammern (wenn man Berichte in der Produktion durchschaut)
- Plastikteile von den Produktionsmaschinen wie auch Verpackungen
Um dies präventiv zu verhindern, bestehen viele Prozessabläufe, Hygieneregeln, Arbeitsanweisungen etc. Dennoch kann es mal passieren, dass bspw. eine lose Schraube in den Lebensmittelproduktionsprozess kommt. Die Lösung: ein Metalldetektor.

Metalldetektoren in der Lebensmittelproduktion
Der Metalldetektor stellt also sicher, dass keine lebensmittelsicherheitsrelevanten Fremdkörper zum Konsumenten gelangen können.
Auf dem Bild sieht man einen Metalldetektor und nebenan drei Teststäbe in den Farben Grün, Rot und Blau mit jeweils einer Grösse von 2,5 mm.
Nachfolgend ging Camilla darauf ein, wie man so ein Gerät validiert und verifiziert.

Die Validierung des Metalldetektors
Beim Metalldetektor wird nicht nur überprüft, ob Metall vorhanden ist. Es ist ein 4-stufiger Prozess damit verbunden, der validiert wird:
- Die Lebensmittel gelangen mit einem Förderband in den Metalldetektor. Ist der Messsensor beim Eingang korrekt eingestellt?
- Der Metalldetektor prüft die Ware. Ist die Sensitivität korrekt eingestellt?
- Die Ware, die Metallteile enthält, wird in einem separaten Behälter ausgeworfen. Funktioniert der Auswurfmechanismus und stimmt die Geschwindigkeit des Förderbands?
- Wenn der Behälter mit der mangelhaften Ware voll ist, wird die Produktion unterbrochen. Funktioniert das?
Wenn nun ein Parameter verändert wird, wie z.B. die Geschwindigkeit des Förderbands, dann verändern sich die Rahmenbedingungen. Und dann muss – wie vorher von Boris erklärt – eine Re-Validierung gemacht werden.

Die Verifizierung des Metalldetektors
Die Verifizierung des Metalldetektors wird bei Delica AG täglich mit den folgenden drei Punkten gemacht:
- Detektiert der Metalldetektor Metallstücke korrekt?
- Funktioniert der Auswurfmechanismus für die mangelhafte Ware?
- Unterbricht die Anlage die Produktion, wenn die Auswurfbox voll ist?
So haben wir nochmals ein anschauliches Beispiel erhalten, wo der Unterschied von Validieren und Verifizieren ist.
Danach folgte eine Frage-Antwort-Runde, in der Boris und Camilla spezifische Fragen der Teilnehmenden beantworteten.
Zum Schluss noch ein paar Tipps
Ganz zum Schluss gab uns Boris noch folgende Tipps:
- Erfahrungsgruppen bilden für einen regelmässigen Austausch
- Weiterbildungen besuchen
- Netzwerk auf- und ausbauen, um aus der Praxis zu lernen
Auch wenn man nicht in einem so stark regulierten Markt tätig ist und keine gesetzlichen Normen erfüllen muss, können Prozessvalidierungen sinnvoll und hilfreich sein. Deshalb hier ein Weiterbildungs-Tipp: Im nächsten Jahr gibt es bei der SAQ-QUALICON einen Grundlagenkurs zur Prozessvalidierung für Unternehmen, die nicht im Medtech- oder Lebensmittel-Bereich tätig sind und keine gesetzlichen Normen erfüllen müssen – aber sich dennoch stetig verbessern möchten bzw. Prüf- und Fehlerkosten senken möchten.
Wenn Du die PowerPoint-Präsentation downloaden möchtest, dann klicke hier auf diesen Link. Weitere QQ-Impulse findest Du jeweils auf unserer Webseite bei den Events.
Haben wir Dich neugierig gemacht? Möchtest Du noch mehr wissen? Nachfolgend haben wir ein paar Weiterbildungen für Dich zusammengestellt:


