
Reinheit nach EN ISO 19227 ist ein zentrales Thema bei der Herstellung von Medizinprodukten. Die Norm trägt zwar den Titel «Reinheit von orthopädischen Implantaten», wird aber für Medizinprodukte aller Klassen als Basis verwendet. Was ist zu beachten, wenn Du ein Sauberkeitskonzept erstellst? Lucio D’Ambrosio gab am QQ-Impuls einen lehrreichen Überblick.
Über eine halbe Million Medizinprodukte sollen sich laut Bundesverband Medizintechnologie (BV-Med) derzeit auf dem europäischen Markt befinden: von Kontaktlinsen und Pflastern über Spritzen und chirurgische Instrumente bis hin zu Implantaten und Herzschrittmachern. Für einen sicheren Einsatz müssen die Hersteller nachweisen, dass sie die Risiken im Herstellungsprozess kennen und im Griff haben, damit keine Gefahr für den Patienten besteht. Die Sauberkeit von Medizinprodukten und deren Verpackungen ist dabei zentral.
Wieso ist Sauberkeit so wichtig für Medizinprodukte?
Seit den 1990er-Jahren hatten sich in Europa die Vorschriften für Sauberkeit von Medizinprodukten und In-vitro-Diagnostika wenig geändert. Man setzte auf die Eigenverantwortung der Hersteller. Doch einige kritische Vorfälle erhöhten den Druck der Öffentlichkeit auf die Politik, den Medizinproduktebereich strenger zu regulieren.
Um die Patienten besser zu schützen, müssen für Medizinprodukte und deren Verpackungen nun Sauberkeitskonzepte etabliert werden.

Sauberkeit ist nicht gleich Sauberkeit
Eine saubere Küche riecht frisch nach Zitrus und ein sauberes Auto glänzt schön in der Sonne. Aber sauber im Sinne der Anforderungen in der Medizintechnik ist das noch lange nicht. An einem sterilisierten Gegenstand können zum Beispiel Öl-Rückstände vorhanden sein, welche bei der Sterilisation nicht entfernt wurden. Standards für Sauberkeit in der Medizintechnik werden in der Norm ISO 19227 definiert.
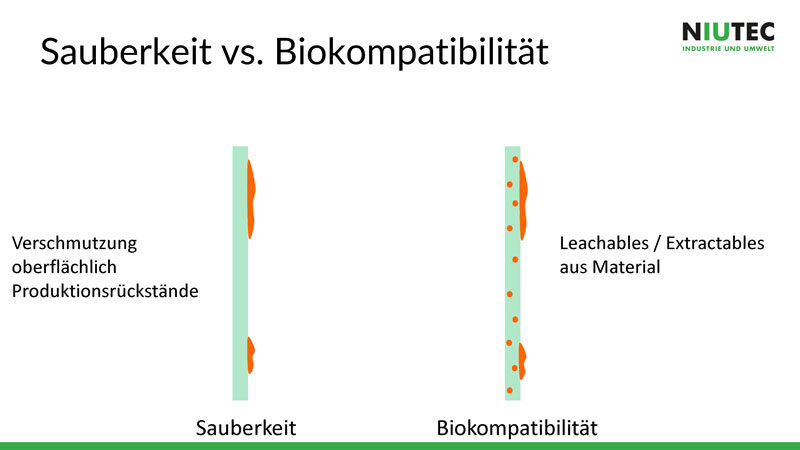
Grundsätzlich gilt es, Sauberkeit und Biokompatibilität zu unterscheiden. Bei der Sauberkeit wird die Verschmutzung der Oberfläche genauer betrachtet. Biokompatibilität bedeutet, dass das Material eines Produkts, zum Beispiel eines Implantates, keine negativen Auswirkungen auf den Stoffwechsel des menschlichen Körpers hat. Darum muss bereits im Entwicklungs- und im Produktionsprozess die Sauberkeit einbezogen werden. Je besser diese ist, desto einfacher wird es in den folgenden Schritten. Und man verhindert dadurch unnötige Kosten und allenfalls Imageschäden, die durch Rückrufaktionen entstehen.
Gefahr durch Herstellungsrückstände
Bei der Herstellung der Produkte werden verschiedene Fertigungsverfahren eingesetzt. Bei jedem dieser Verfahren verbleiben auf den Oberflächen partikuläre und/oder filmisch-chemische Rückstände, wie zum Beispiel Trennmittel, Silikone, Späne, Staub, Abrieb, Strahlmittel, Reinigungsmittel, Klebstoffe, Talkum von den Handschuhen, Abtrieb vom Verpackungsmaterial oder Keime. Diese Kontaminationen stellen je nach Risikoklasse, in die ein Medizinprodukt eingestuft ist, ein unterschiedlich grosses Schädigungspotenzial für Patienten dar. Das heisst, um die Sauberkeit der Medizinprodukte zu gewährleisten, ist die Reinigung ein Prozess entlang der gesamten Fertigungskette.
Sauber von A bis Z mit dem Sauberkeitskonzept
Bei der Vorbereitung für ein Sauberkeitskonzept ist zu berücksichtigen, aus welchem Material das Produkt gefertigt wird und welche potenziellen Verschmutzungen im Produktionsprozess wie auch in den weiteren Prozessen vorkommen können.
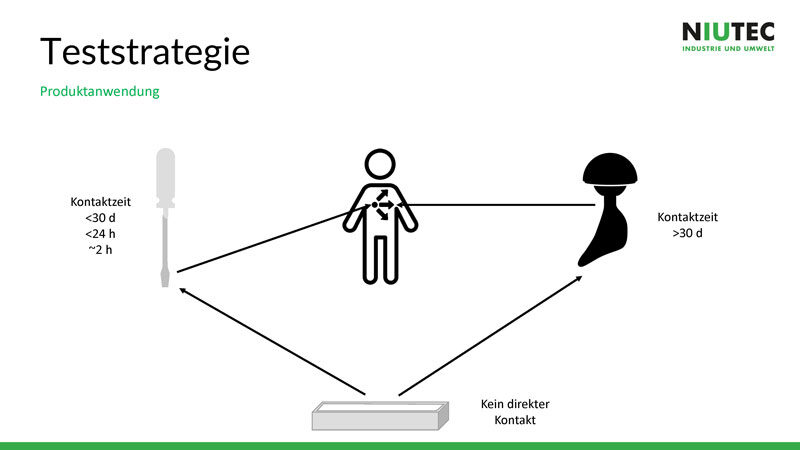
Für die Teststrategie ist die Produktanwendung ausschlaggebend, wie sauber das Produkt sein muss und wie genau getestet werden muss. Hat das Produkt direkten Patientenkontakt, kommt es nur kurz mit dem Gewebe des Patienten in Kontakt oder ist es gar ein bleibendes Implantat? Diese Fragen sind im Vorfeld unbedingt abzuklären.
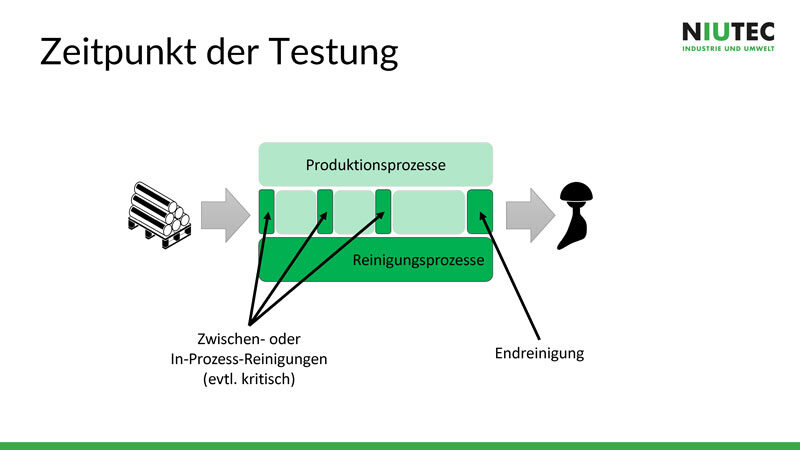
Wenn Material und Einsatz des Produktes geklärt sind, wird definiert, wann und wie oft die Sauberkeit getestet wird. Eine Endreinigung reicht in den meisten Fällen nicht aus, da am Schluss nicht mit für den Patienten schädlichen Stoffen gereinigt werden darf, aber zu wenig aggressive Mittel gewisse Verschmutzungen nicht entfernen können. Darum müssen während des Produktionsprozesses sinnvolle Zwischen- oder In-Prozess-Reinigungen eingeplant werden, bei denen Chemie mit Chemie gereinigt wird. Bei der Endreinigung hingegen wird in der Regel mit Wasser gereinigt.

Um festzulegen, welche Verschmutzung wann wie am besten entfernt werden kann, ohne unnötig viele Reinigungsdurchläufe machen zu müssen, empfiehlt es sich, alle eingesetzten Stoffe risikobasiert unter die Lupe zu nehmen. Dabei geht es einerseits darum, wie einfach diese zu reinigen sind und andererseits um die Vermischungen unterschiedlicher Betriebs- und Hilfsstoffe, die zu Problemen bei der Reinigung führen kann. Idealerweise wird versucht, kritische Stoffe durch eine Prozessoptimierung aus der Fertigung zu eliminieren sowie die Zahl und Menge der eingesetzten Betriebs- und Hilfsstoffe auf ein Minimum zu begrenzen. Hierfür muss eine Einteilung in Verschmutzungskategorien gemacht werden, um zu wissen, welche potenziellen Verschmutzungen da sind und in welcher Reihenfolge diese zu beseitigen sind. Die richtige Kategorisierung ist die notwendige Basis, um die Reinheit zu gewährleisten: Für jede Verschmutzung gibt es ein passendes Lösungsmittel.

Die ISO-Norm 19227 ist aus dem Bereich der Implantate, hat sich aber in der Praxis als Basis für alle weiteren Reinigungen etabliert.
Neben sichtbaren Verunreinigungen gilt es, Kontaminationen durch Mikroorganismen zu erkennen wie auch Kontaminationen organischer und anorganischer Art sowie Partikelrückstände.
Die Gratwanderung mit den Grenzwerten
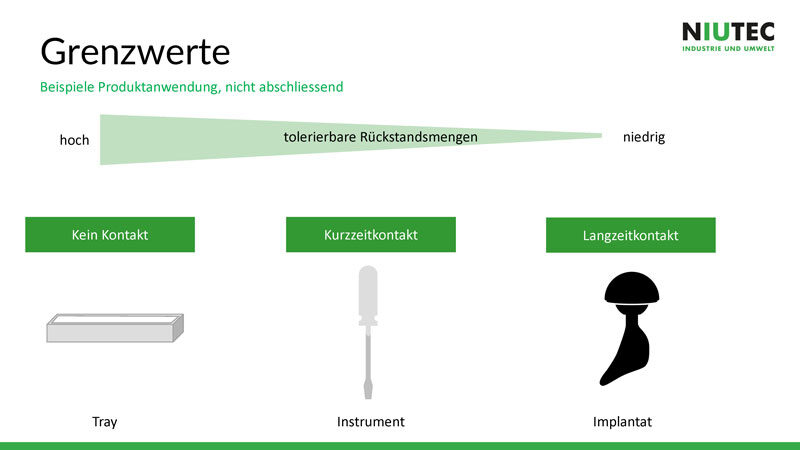
Es gibt keine definierten Grenzwerte für Rückstandsmengen an Verpackungen, Werkzeugen oder Implantaten. Um Grenzwerte zu definieren, müssen folgende Punkte berücksichtigt werden:
- Wie lange kommt ein Produkt in Kontakt mit dem Patienten?
- Wie gross ist die Fläche des Produkts, die mit menschlichem Gewebe in Kontakt kommt?
Folglich haben Implantate niedrigere Grenzwerte als die Produkte, die kurzen oder gar keinen direkten Kontakt mit den Patienten haben.

Auf
der Folie sieht man Beispiele, an denen man sich
orientieren kann. Für die Definition einiger Grenzwerte existieren Normen und
Guidelines, viele Grenzwerte müssen jedoch individuell definiert werden. Die
Grenzwerte werden in der Regel in Zusammenarbeit mit dem Labor, Toxikologen und
dem Inverkehrbringer bzw. dem Zulieferer erarbeitet.
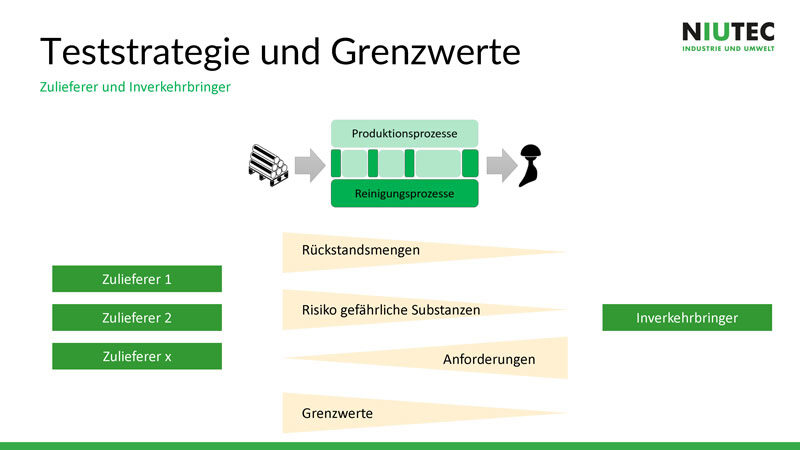
Am Anfang des Prozesses sind immer mehr Verschmutzungen bzw.
Rückstände vorhanden. Je weiter der Produktionsprozess fortgeschritten ist, je
reiner wird das Produkt mit dem Ziel, dass bis zur Inverkehrbringung das
Produkt komplett rein ist. Das heisst, je weiter der Produktionsprozess
fortgeschritten ist, je höher werden die Anforderungen und je niedriger sind
die Grenzwerte für Kontaminationen. Die Rückstand- bzw. Verschmutzungsmenge und
das Risiko gefährlicher Substanzen nimmt dagegen immer weiter ab.

Ein Beispiel für eine Definition von Grenzwerten

Alarmwerte definieren
Eine Alarmierung, wenn der Grenzwert bereits überschritten ist, ist viel zu spät und kann teuer werden: Das Unternehmen muss die Produktion stoppen und schauen, wie der Grenzwert wieder unterschritten werden kann. Ein unterhalb des Grenzwertes angesetzter Alarmwert hilft, entsprechende Massnahmen zu ergreifen, damit die Produktion nicht ins Stocken kommt. Mit einem fortlaufenden Monitoring und einem definierten Alarmwert bei 40 bis 60% des Grenzwerts kann sichergestellt werden, dass der Grenzwert nicht überschritten wird.
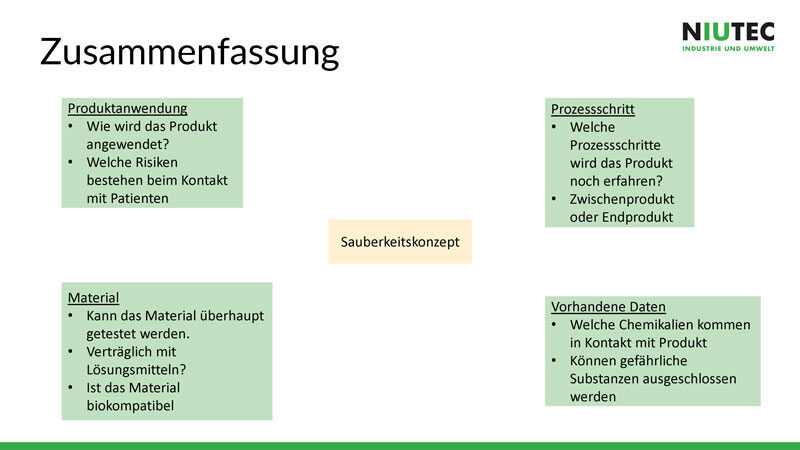
Schritt für Schritt zum Sauberkeitskonzept
Zusammengefasst wird ein Sauberkeitskonzept in den folgenden vier Schritten erstellt:
- Produktanwendung eingrenzen und beschreiben
- Material analysieren
- Reinigungs- Prozessschritte festhalten
- Vorhandene Daten prüfen und hinzuziehen
Da es keine fix festgelegten Grenzwerte gibt, geht es vor allem darum, bei Behörden und Regulatoren nachweisen zu können, dass man sich mit den Risiken bezüglich Sauberkeit auseinandersetzt und fachlich fundierte Grenzwerte definiert hat. Dass dies dokumentiert und laufend überwacht wird, versteht sich von selbst. Denn nur, wenn die produzierten Medizinprodukte eine saubere Sache sind, dienen sie dem Wohl des Patienten und sind Basis für den Erfolg des Unternehmens.

Datenvisualisierung
Video QQ-Impuls
Wir haben Dir den gesamten QQ-Impuls aufgezeichnet, so dass Du Dich auch im Nachhinein über dieses interessante Thema informieren kannst.
Lerne noch mehr zum Thema!



Text: Anja Zell